Artículos de Investigación
FLC
Fighting rare cancers: lessons from fibrolamellar hepatocellular carcinoma.
- Es de 2023-3.
- Cuenta primero la historia del descubrimiento de la causa del tumor y como la presencia del gen de fusión DNAJB1-PRKACA es común, pero no siempre es el caso.
- El tamaño del tumor en el momento de la resección es irrelevante de cara al pronóstico. Que lo relevante es cuántas metástasis se observen. Y que esta no es mayor por ser más grande. Y que no se sabe por qué unos metastatizan más que otros.
- De hecho, la hija del autor del artículo seguía viva 14 años después de la resección.
- Las mujeres sobreviven al FLC más tiempo que los hombres.
- Igualmente que, aunque parezca un tipo de cáncer con una causa muy concreta, la sensibilidad a los tratamientos varía mucho de unos pacientes a otros. Tampoco se sabe por qué.
- Dice que es común diagnosticar FLC equivocadamente.
- Dice que el hígado es responsable de la eliminación del 70% del amoniaco del cuerpo; por ello, en caso de fallo hepático el amoniaco se acumula y produce una encefalopatía. Pero añade que, extrañamente, este es un problema común en pacientes con FLC pese a que la mayor parte de su hígado sea funcional.
- Encontraron algún fármaco quimioterapéutico que funciona in-vitro: irinotecan (que bloquea a una enzima implicada en la replicación y la transcripción del ADN) o navitoclax (que desbloquea rutas de apoptosis, pero que geneta toxicidad plaquetaria).
- Muestra que sin la proteína mutante DNAJB1-PRKACA (cuya producción bloquearon con terapia de ARNsh en células cultivadas), las células tumorales mueren: esto es porque la kinasa A, hiperactiva o no, es necesaria para la supervivencia de las células.
Dice que hay variantes de FLC donde no está presente la proteína DNAJB1-PRKACA (la kinasa A mutante hiperactiva); en este caso el tumor podía ser causado por:
- la inactivación de un gen (p.e. BAP1) que da como resultado la ausencia de una molécula que modula la actividad de la kinasa A, por lo que el resultado final sigue siendo una kinasa A igualmente hiperactiva, o
- por otras fusiones de genes (p.e. ATP1B1-PRKACA) con el mismo resultado final: kinasa A hiperactiva. En todos estos casos, ni una vacuna de péptidos ni una terapia con ARNi específicas contra la proteína/gen DNAJB1-PRKACA pueden funcionar.
Por ello, antes de una terapia de ese tipo, es necesario caracterizar cuál es la causa genética y bioquímica exacta en cada paciente.
Por su parte una terapia tal como un combinado de ICIs podría ser útil en todos los casos que se deban a proteínas de fusión (sea DNAJB1-PRKACA, ATP1B1-PRKACA u otra) ya que estas sirven como neoantígenos reconocibles por los linfocitos T, pero probablemente no funcione cuando la causa sea la inactivación de un gen que codifique para un modulador de la kinasa A, ya que en este caso no habría nada que pudiera funcionar como neoantígeno.
Surprising origins for a rare cancer.
- Es de 2024-6.
- Las enzimas tienen un centro activo en su dominio catalítico, que es por donde se unen al sustrato y con el que, por tanto, efectúan su acción. Pero también han de tener un mecanismo para ser frenadas cuando convenga a la célula (por ejemplo, para impedirles el paso al núcleo). En el caso de la kinasa A normal este mecanismo se llama "inhibición no competitiva" y consiste en que tienen un dominio al que se unen las moléculas inhibidoras; al hacerlo cambia la forma del centro activo y la enzima ya no se puede unir a sus sustratos.
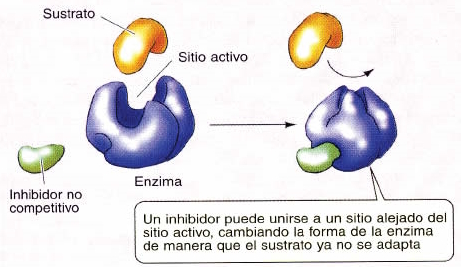
- La kinasa A del FLC debido a genes de fusión (sea DNAJB1-PRKACA, ATP1B1-PRKACA u otra) ha perdido este dominio y no puede ser inhibida: campa a sus anchas por la célula, sin freno para así fosforilar a cualquier molécula que encaje en su centro activo, desregulando por completo el funcionamiento de la célula.
- Los mismos efectos desreguladores suceden si la kinasa A no mutante está sobreexpresada, lo que sucede cuando el FLC se debe a la inactivación de un gen (p.e. BAP1) o cuando se incrementa artificialmente la presencia de kinasa A en un hepatocito.
- Es decir, el FLC se debe bien a una kinasa A no inhibible, bien a una kinasa A muy abundante, en todos los casos a causa de mutaciones genéticas, siendo la más común la que da lugar al gen de fusión DNAJB1-PRKACA.
- Un posible enfoque terapéutico derivado de esto sería buscar cómo inhibir a la kinasa A mutante aprovechando el dominio de la chaperona (DNAJB1) que tiene o la zona de fusión entre ambos dominios. No mucho, no nuevo y no fácil.
- Es de 2024-3.
- Encontraron que no todos, pero sí la mayoría (20/24) de los pacientes expresan neoantígenos de la proteína de fusión en sus células tumorales, pero que expresan pocos, como es común en tumores con una única proteína mutante recurrente.
- Con todo, lo anterior permitiría que las células tumorales fuesen identificables y eliminables por el S.I. en estos pacientes.
- Y así encontraron que en los tumores de FLC hay linfocitos infiltrados (pero pocos) y que entre ellos hay linfocitos T citotóxicos capaces de reconocer a los neoantígenos de las células tumorales y matarlas (pero pocos), porque en su superficie tienen TCRs que reconocen a los neoantígenos (pero pocos). Es decir, en concordancia con los limitados neoantígenos expresados, hay respuesta inmunitaria, pero débil.
- En concreto un clon de linfocitos T citotóxicos era capaz de reconocer a uno de los péptidos de laboratorio hechos a partir de la zona de fusión de la proteína mutante DNAJB1-PRKACA; este tenía 10 aminoácidos, 9 eran de la zona DNAJB1 y 1 era de la zona PRKACA. La respuesta in vitro y en ratones de esos linfocitos T contra las células tumorales era muy eficaz.
- Si el gen que codificaba para el TCR (receptor) que permitía a esos linfocitos T reconocer al neoantígeno, era transferido a otros clones de linfocitos T citotóxicos, estos también se volvían capaces de identificar y matar a las células tumorales.
- Todo lo anterior viene a decir que:
- La mayoria de las personas tenemos ya en el cuerpo clones de linfocitos T capaces de reconocer y atacar a las células FLC, porque la mayoría presentan neoantígenos reconocibles como tales.
- Cabe deducir que si esos clones de linfocitos T útiles contra el tumor están presentes pero no se activan es porque el tumor es muy eficiente manteniéndolos lejos a base de desactivar al que se acerca (que ya no da la voz de alarma para atraer a otros), por lo que un tratamiento con ICIs parece necesario si el enfoque es la inmunoterapia.
- Pero también dice que se ha visto una correlación positiva entre la eficacia de un tratamiento de cáncer con ICIs y la presencia de TILs con muchos TCRs específicos. Aquí escasean porque las células tumorales tienen pocos neoantígenos, lo que da lugar a una respuesta inmunitaria débil y el tratamiento con ICIs tiene un éxito desigual: quitarle el bozal a los perros de presa tiene sentido cuando hay muchos y la identifican bien, lo que no sucede aquí. Hay que aumentar y enfadar a la jauría, y es por esto que la inmunoterapia contra el FLC se podría beneficiar mucho de cualquier tratamiento que genere muchos linfocitos T anti-FLC: vacuna de neoepítopos (fragmentos peptídicos del neoantígeno, la proteína de fusión) o linfocitos CAR-T anti-FLC.
- Es decir, que la inmunoterapia debería de combinar ICIs con vacuna o CAR-T.
- Que no todos los péptidos de la zona de fusión de la proteína mutante valen como neoepítopos.
CDK7 is a novel therapeutic target in fibrolamellar carcinoma.
- Es de 2025-12.
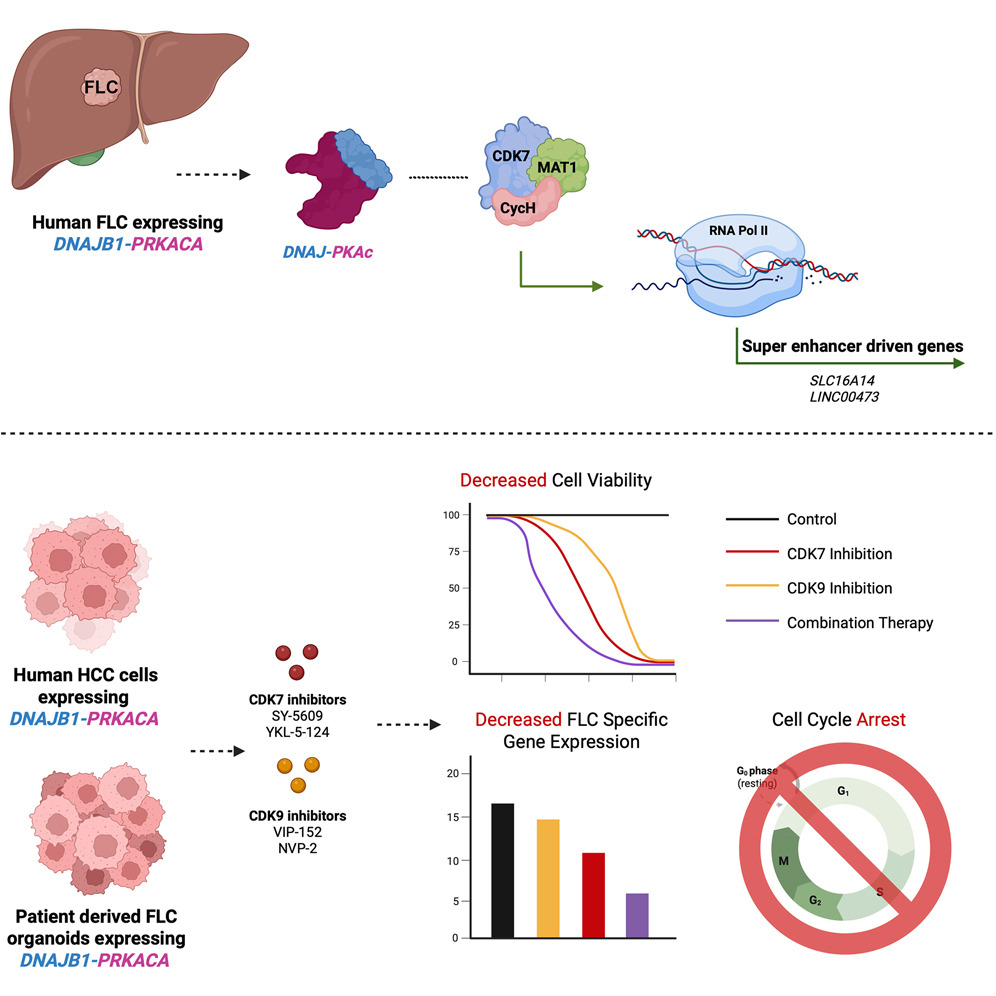
- Encontraron que un posible mecanismo por el que la kinasa A de fusión favorece que las células FLC se tornen tumorales seria favoreciendo la sobreexpresión de genes tales como como SLC16A14 y LINC00473, que son decisivos en el desarrollo del FLC.
- Se encontró que en las células FLC (al igual que en otras clases de tumor) hay genes reguladores de los oncogenes anteriores sobreactivados; que esto es porque la RNA-pol II (la enzima que transcribe estos genes) está sobreactivada en estos genes reguladores; que esto está en relación con el hecho de estar fosforilada en un lugar concreto; que esta fosforilación es debida a la actividad de la quinasa dependiente de ciclinas CDK7 (que además de promover la transcripción del ADN, promueve la división celular al inducir el avance a la fase S del ciclo celular); que la enzima CDK7 está sobreactivada en células FLC y que esto se debería a la actividad de la kinasa A mutante.
- Y así se encontró (in vitro y en organoides) que la inhibición de la kinasa CDK7 mataba a las células FLC, dejando intactas a los demás hepatocitos. Y que si además se inhibía a la kinasa CDK9, los efectos eran aún mayores.
- Esto convierte a las kinasas CDK7 y CDK9 en dianas quimioterapéuticas contra el FLC.
Silenciamiento de genes
- Es de 2024-3.
- Los ARNs largos no codificantes (long non-coding RNA, lncRNAs) son ARNs que resultan de la expresión de genes reguladores. Los ARNlnc, por tanto, no están destinados a ser traducidos a proteínas en los ribosomas (aunque algunos sí se traducen a micropéptidos). Una de sus funciones es activar o desactivar a genes codificantes concretos.
- Encontraron que el ARNlnc llamado LINC00473 estaba sobreexpresado en las células FLC en una medida que se correspondía con la actividad de la kinasa mutante responsable del FLC. Este ARNlnc está también sobreexpresado en ciertos tumores de pulmón, pero subexpresado en tumores colorrectales.
- Encontraron que la sobreexpresión del ARNlnc LINC00473 daba lugar a la sobreexpresión de genes que inhibían la apoptosis y de genes que favorecen la generación de energía en las mitocondrias.
- En consecuencia, el ARNlnc LINC00473 puede ser usado como:
- marcador de la actividad de la kinasa A mutante;
- diana de posibles tratamientos terapéuticos basados en "silenciamiento de genes", bien con CRISPR, bien con ARN de interferencia.
Quimioterapia
Neurotensin as a source of cyclic AMP and co-mitogen in fibrolamellar hepatocellular carcinoma.
- Es de 2019-8.
- Dice que la kinasa A mutante del FLC, la proteína que desregula los hepatocitos y los vuelve tumorales, es dependiente del AMP cíclico para funcionar, tal como lo es la kinasa A normal. Y que por lo tanto las vías metabólicas que resultan en la producción de AMPc han de estar sobreactivadas en las células FLC.
- Y ese es el caso: una vía metabólica que usa neurotensinas para generar AMPc está sobreactivada en células FLC.
- Por consiguiente inhibir a las neurotensinas reduciría la actividad de la kinasa A y reduciría el ritmo de mitosis en células FLC, frenando el desarrollo del tumor.
- Los inhibidores de neurotensinas funcionarían pues como los inhibidores de kinasas. Y parece difícil que no tuvieran efectos secundarios generalizados, ya que estas enzimas son ubicuas.
ICIs
- Es de 2022-10.
- Es un metaestudio que dice que de 19 pacientes que recibieron tratamiento con ICIs (17 de ellos 1; 2 de ellos 2), 4 de ellos en combinación con otros tratamientos, sólo 3 (14%) mostraron mejoras clínicas parciales.
- Se piensa que la baja respuesta al tratamiento con ICIs se podría deber (a) a que la baja carga mutagénica (TMB) de los tumores FLC, que se manifiesta en una expresión escasa de neoantígenos, tiene poca capacidad de enfadar a los linfocitos T, y (b) a que las células tumorales FLC no expresan PD-L1, por lo que el problema (el que las células FLC evadan la acción de los linfocitos T) no consiste en la actuación de ICs, y por tanto no se puede atajar con ICIs.
- Es de 2022-12.
- Dice que una paciente en los 30 que había sufrido sucesivas recidivas tras cirugías y previos tratamientos, incluido el tratamiento con ICI de tipo anti-PDL1 (atezolizumab ) ha logrado una curación completa con un tratamiento combinado con ICIs de tipo anti-PD1 (nivolumab) y anti-CTLA4 (ipilimumab), pese a que tratamiento hubo de ser interrumpido durante 12 semanas o más por diarreas y prurito excesivos, lo que provocó una recaída.
- El tratamiento fue predicho por el análisis molecular y genético del tumor ENLIGHT llevado a cabo por Pangea Biomed. El artículo ha sido escrito por científicos de esa empresa y es un tratamiento que también se puede predecir sin test alguno (ver abajo).
Using dual checkpoint blockade to treat fibrolamellar hepatocellular carcinoma.
- Es de 2020-2.
- La paciente es una mujer de Munich de 22 años con metástasis avanzadas, tratada previamente con quimioterapia. Tras un tratamiento combinado con ICIs de tipo anti-PD1 (nivolumab) y anti-CTLA4 (ipilimumab) logró una curación casi completa a los 3 meses de iniciar el tratamiento. 1 año después estaba limpia.
Vacunas de péptidos
- Es de 2022-10.
- Dice que un paciente vacunado en el Hospital Universitario de Tubinga con péptidos derivados de la proteína DNAJB1-PRKACA (vacuna FusionVAC-22), en combinación con inhibidores de PARP, ha conseguido inducir la reactivación de los linfocitos T auxiliares y los T citotóxicos específicos contra células FLC en un paciente que había sufrido sucesivas recidivas tras previos tratamientos de cirugía y radioterapia, y mantenerlo libre de recaídas 21 meses después del inicio de la vacunación (parece que 3 años, ya).
- En base a esto han iniciado un ensayo clínico de la vacuna en combinación con atezolizumab (ICI de tipo anti-PDL1) [ver abajo].
- Es de 2023-11.
- Son los resultados preliminares del ensayo clínico de la vacuna de péptido + Nivolumab (ICI anti-PD1) + Ipilimumab (ICI anti-CTLA4) de la Universidad Johns Hopkins, Baltimore [ver abajo].
- Se estudió a 16 pacientes (sólo 3 mujeres), con metástasis progresivas incontrolables por otras terapias, con presencia confirmada del gen DNAJB1-PRKACA, sin tratamiento previo con ICIs y sin tratamientos adicionales durante el ensayo.
- Algunos pacientes sufrieron reacciones autoinmunes, que pudieron ser manejadas, además de reacciones en la zona de inyección, dolores de cabeza y fatiga.
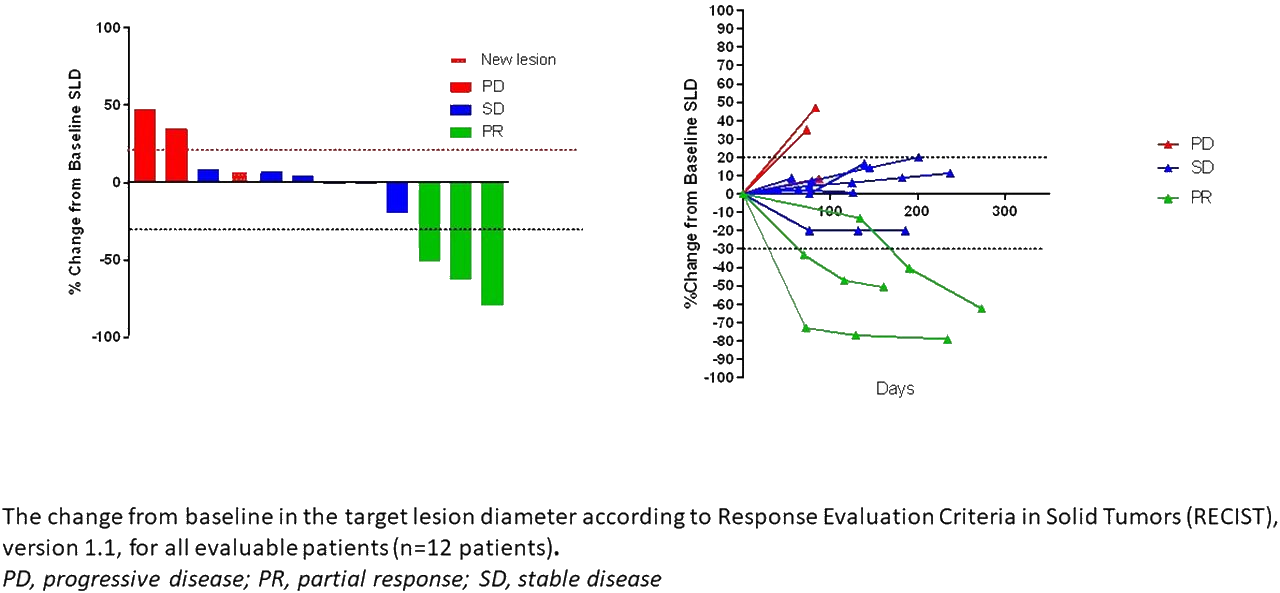
- De los 12 pacientes finalmente evaluables:
- 3 vieron como su enfermedad seguía progresando;
- 9 generaron una respuesta inmunitaria incrementada contra el tumor:
- 6 vieron sus tumores estabilizados o algo reducidos ("enfermedad controlada");
- 3 tuvieron una reducción de sus tumores superior al 30% ("respuesta parcial");
- ninguno alcanzó la "remisión clínica".
- Es de 2024-7.
- Los ARNs largos no codificantes (long non-coding RNA, lncRNAs) son ARNs que resultan de la expresión de genes reguladores. Los ARNlnc, por tanto, no están destinados a ser traducidos a proteínas en los ribosomas, aunque algunos sí se traducen a micropéptidos.
- Un problema en carcinoma hepatocelular (HCC) es que las células tumorales apenas expresan neoantígenos.
- Estos investigadores encontraron que de los neoantígenos expresados por las células HCC, el 40% eran micropéptidos codificados por ARNlnc. 2 de ellos inducían respuesta inmunitaria en ratones con xenoinjerto y serían buenos candidatos para ser utilizados en vacunas contra el HCC.
Glosario
Genética
Genes
En nuestro ADN sólo el 4% son genes. Y sólo el 2% son genes que se transcriben a ARN mensajero (ARNm) y luego se traducen a proteínas en los ribosomas. Estos son los genes codificantes, y son los que determinan cómo eres.
El otro 2% codifica para ARN ribosómico o de transferencia (que ayudan en la traducción) o son genes que regulan la expresión de los genes codificantes.
Ese 2% de ADN que codifica para proteínas es un total de 20.400 genes.
Los genes de todos los seres vivos (salvo los de las bacterias) tienen, a su vez, regiones codificantes, llamadas exones, intercaladas por regiones no codificantes, llamadas intrones. Esto es muy útil para, por recombinación de exones, generar una diversidad de proteínas muchísimo mayor que 20.400 clases. Eso sí, los intrones han de ser extirpados del ARNm antes de que este llegue a los ribosomas para ser traducido a proteína. Esto se hace en el núcleo mediante un proceso llamado "corte y empalme" (splicing).
Transcriptoma
Todas las células de una persona tienen los mismos 20.400 genes codificantes. Y tenemos entre 200 a 2000 tipos celulares (según como se cuenten), por lo que lo que las diferencia no son los genes codificantes que tengan sino los que se expresan: el gen de la lactasa no se expresa en una célula de la oreja, por ejemplo.
Para saber cuáles son los genes codificantes que se expresan en un tipo celular lo que se hace es buscar el ARN mensajero (ARNm) que haya en el citoplasma de esa célula, ya que es el resultado de la expresión de aquellos, y se secuencia. Eso es el transcriptoma. Es la serie de genes codificantes para proteínas que se expresan en una célula concreta, y por lo tanto lo que la caracteriza a nivel genético.
Identificar el transcriptoma de una célula es muy caro. Pero es más rápido y útil que secuenciar su genoma completo, ya que...
- se evita secuenciar la parte del ADN que no son genes codificantes (un 98%);
- se evita secuenciar los genes de esa célula que no se expresan en ella;
- se evita secuenciar los intrones (secuencias no codificantes) que hay intercaladas en los genes, ya que se extirpan del ARNm antes de que salga del núcleo al citoplasma.
- se detecta qué genes se están sobreexpresando y qué genes se están subexpresando en la célula tumoral, los cuáles podrían servir como dianas terapéuticas.
FLC
DNAJB1-PRKACA
El gen PRKACA codifica para la kinasa A. Las kinasas son enzimas que transfieren grupos fosfato de una molécula a otra; esta última es típicamente otra enzima que, al recibir el grupo fosfato pasa de inactivada a activada. Las kinasas, por lo tanto, son enzimas con la tarea genérica de activar a otras enzimas con tareas más específicas, que den como resultado final acciones tales como la angiogénesis, el crecimiento celular, la mitosis, o cualquier otra.
A su vez las kinasas han de activarse para realizar su tarea; típicamente esto es así: se presentan insertas en la membrana de una célula, y se activan cuando una molécula mensajera (una citoquina o una hormona) emitida por otra célula se une a ellas.
El gen DNAJB1 codifica para una chaperona, que es una enzima que también sirve para activar a otras proteínas; en concreto hace la tarea esencial de ayudar a plegar correctamente el péptido recién fabricado en el ribosoma para que este pase a ser una proteína con la forma exacta que le permita desempeñar su función, a falta de combinarse con las piezas moleculares que pueda necesitar adicionalmente (un grupo fosfato, un ion magnesio, un grupo hemo, una vitamina...)
El gen de fusión, mutante, DNAJB1-PRKACA, se forma por una mutación génica que consiste en la delección (pérdida) de 8 genes ubicados entre los genes PRKACA y DNAJB1. También se pierden 1 exón del primero y 2 exones del segundo. De este modo, lo que queda de ambos genes queda combinado en un único gen de fusión anómalo, que se transcribe a un ARNm de fusión anómalo, cuya traducción en los ribosomas da lugar a una proteína de fusión anómala.
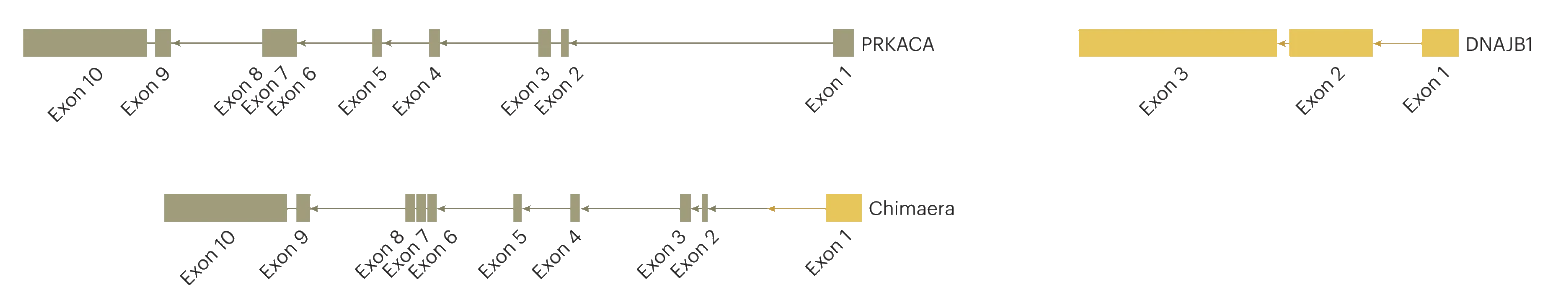
Esta proteína de fusión es la kinasa A codificada en condiciones normales por el gen PRKACA, pero que ahora es anormalmente hiperactiva. A veces se llama la "proteína DP". Su hiperactividad es la que da como resultado el comportamiento tumoral de las células mutantes.
TMB
Es la carga mutacional del tumor. En un tumor normal, de rápido crecimiento, las células suelen presentar más mutaciones de lo normal, ya que estas normalmente se producen más por errores en la replicación del ADN previa a la mitosis que por agentes mutágenos externos: a más tasa de división celular, más mutaciones.
Pero los tumores FLC son de crecimiento lento y no suelen acumular más mutaciones que la que resulta en la proteína de fusión DNAJB1-PRKACA. Esto hace que los análisis genéticos de estos tumores tengan pocas posibilidades de mostrar alguna diana terapéutica más allá de la proteína mutante.
También resulta en que las células FLC normalmente sólo expresen neoantígenos (más bien neoepítopos) derivados de la proteína mutante (y además no muchos). Por eso los tumores FLC presentan pocos linfocitos (TILs) anti-FLC y un tratamiento sólo con ICIs no suele ser suficiente.
TILs
Son los linfocitos infiltrados en el tumor. En el caso del FLC son muy pocos; de entre ellos son muy pocos los que son capaces de reconocer a los neoantígenos de las células tumorales; y los que lo hacen presentan pocos TCRs específicos contra estos. Es decir, la respuesta inmunitaria endógena (natural) contra el FLC es muy débil.
Esto es porque las células FLC presentan muy pocos neoantígenos y por ende tienen poca inmunogenicidad. Además tienen la capacidad de "apagar" a los linfocitos T citotóxicos manipulando sus puntos de control (ICs).
Es por esto que la inmunoterapia contra FLC debe pasar...
- tanto por la multiplicación de linfocitos T que reconozcan a los neoantígenos del tumor (bien inducida por vacunas de péptidos, bien generados in-vitro por CAR-T);
- como por inhibir esa capacidad del tumor de apagar a los linfocitos, que es lo que se trata de conseguir con el uso ICIs.
Sistema Inmunitario
Antígenos, Receptores de antígenos, Linfocitos
Los antígenos son las moléculas extrañas al organismo que son reconocidas como tales por los receptores de antígenos que poseen las células responsables de la inmunidad adaptativa, es decir, los linfocitos.
Los antígenos pueden ser moléculas libres (como las exotoxinas producidas por Chlostridium) o asociadas a la superficie de virus o de células. Las células que pueden presentar antígenos son todas las extrañas al organismo, como las de bacterias, protozoos, hongos, injertos o transplantes. Pero también las células propias, bien cuando son infectadas por virus, bien cuando se vuelven tumorales, bien cuando llegan a la senescencia, suelen expresar proteínas nuevas, extrañas; estas se llaman neoantígenos porque son antígenos de nueva formación.
Las zonas de una molécula de antígeno a las que se unen los receptores que hay en los linfocitos se llaman epítopos. Una molécula antigénica puede tener un solo epítopo o más. Cuantos más tenga, más clases diferentes de linfocitos la reconocerán y la respuesta inmunitaria será más enérgica.
Los linfocitos son las células humanas que reconocen a los antígenos de forma específica: cada linaje (clon) de linfocitos está diseñado para reconocer un único tipo de antígeno. En conjunto pueden reconocer unos 10.000.000.000 antígenos diferentes.
Los receptores de antígenos de los linfocitos T se llaman "receptores de antígenos de los linfocitos T" (T-cell receptors, TCRs). Los receptores de antígenos de los linfocitos B se llaman anticuerpos.
Cada TCR se puede unir a una única molécula de antígeno; cada anticuerpo a 2.
Cuando un linfocito T citotóxico (T killer, Tk, T-CD8) reconoce a un antígeno con sus TCR, lo elimina perforando su membrana celular o desencadenando su apoptosis (muerte celular regulada metabólicamente). Los T citotóxicos patrullan continuamente para detectar neoantígenos presentados por células nativas tumorales, infectadas por virus o senescentes. Tras eliminarlas, algunas permanecen como células de memoria.
Cuando un linfocito T auxiliar (T helper, Th, subgrupo de T-CD4) reconoce a un antígeno con sus TCR (normalmente porque se lo presentan, no porque él lo encuentre azarosamente), busca a los clones de linfocitos Tk y linfocitos B que reconocen al mismo antígeno, para activarlos.
Cuando un linfocito B reconoce a un antígeno con sus anticuerpos de superficie y es activado por el linfocito Th correspondiente, se diferencia a célula plasmática que prolifera, y produce y libera masivamente al medio (plasma sanguíneo, linfa, mucus, saliva, lágrimas, leche materna) anticuerpos en forma de monómeros, dímeros, trímeros o pentámeros. Estos tienen el efecto de neutralizar a los antígenos y de marcarlos para que los efectores de la inmunidad innata los eliminen (fagocitos, células NK, proteínas del complemento).
Células presentadoras de antígenos
En la sangre hay varias clases de glóbulos blancos. Una son los monocitos. Estos, como la mayoría de los leucocitos, pueden migrar de la sangre a los tejidos. Cuando lo hacen, los monocitos se diferencian bien a macrófagos, bien a células dendríticas. Ambos tienen doble actividad:
- Fagocítica (dominante en los macrófagos): capturan todo tipo de microorganismos, toxinas, células propias tumorales y células propias infectadas por virus de forma inespecífica, y las digieren en su interior.
- Presentadora de antígenos (dominante en las células dendríticas); algunos fragmentos resultantes de la digestión del m.o. van a servir de antígenos: el fagocito los lleva a su superficie, los asocia a las proteínas HLA tipo II que hay allí, y así expuestos se los presenta a los linfocitos T auxiliares (Th), activándolos contra todo tipo de célula o partícula que lleve ese antígeno en su superficie. De este modo la respuesta inmunitaria pasa de ser genérica (o innata) a específica (o adaptativa), y su agente nuclear pasan a ser los linfocitos Th, los cuales proceden ahora a activar a los clones de linfocitos Tk y B que lleven en su superficie receptores contra ese antígeno concreto (TCRs o anticuerpos, respectivamente).
Proteínas MCH, CMH o HLA
Son lo mismo. Son las proteínas del complejo mayor de histocompatibilidad, codificadas en humanos por los genes HLA. Las de tipo I las tienen todas las células de un individuo (salvo sus glóbulos rojos) y sirven para 2 cosas:
- Como DNI de las células de un individuo, ya que el conjunto de las proteínas HLA de cada persona específico de ella y común a todas sus células; las células que presentan su patrón propio de proteínas HLA no son eliminadas por su sistema inmunitario, las demás sí. Esto sirve para poder identificar y eliminar células ajenas al organismo.
- Para presentar antígenos: una célula continuamente toma proteínas de su citoplasma al azar, las trocea en fragmentos pequeños (péptidos = cadenas simples de aminoácidos) y a estos los expone asociados a las proteínas HLA, que funcionan así como peanas expositoras presentes en la superficie celular. Esto sirve para identificar y eliminar células propias tumorales, infectadas por virus o senescentes, ya que todas ellas suelen expresar proteínas específicas de la que carece una célula normal (y que sirven de neoantígenos cuando son expuestas al exterior). En el FLC es el caso de la proteína DNAJB1-PRKACA. En este caso, la misma proteína responsable del tumor es expuesta a trozos en la superficie de la propia célula tumoral para que los linfocitos T citotóxicos puedan identificarla como célula anómala y eliminarla.
Hay 2 clases de proteínas HLA:
- Las de tipo I: las tienen todas las células nucleadas del organismo; cumplen ambas funciones.
- Las de tipo II: las tienen sólo los glóbulos blancos con función presentadora de antígenos (macrófagos y célula dendríticas, sobre todo); sirven para presentar a los linfocitos Th antígenos de células o partículas exógenas que han sido fagocitadas y desencadenar la respuesta imunitaria adaptativa.
Puntos de control inmunitario
Los puntos de control inmunitario (inmune checkpoints, ICs) son proteínas de superficie de los linfocitos T citotóxicos y T auxiliares que actúan como interruptores que modulan su grado de respuesta una vez que han reconocido a su antígeno. Hay ICs de tipo activador e ICs de tipo inhibidor:
- un IC de tipo estimulador presente en la superficie de los linfocitos T citotóxicos es CD28;
- dos ICs de tipo inhibidor presentes en la superficie de los linfocitos T citotóxicos son CTLA-4 y PD-1.
Para que un linfocito T sea activado es necesario que...
- sus TCRs se hayan unido a su antígeno específico;
- sus ICs estimuladores se hayan unido a sus ligandos (se haya pulsado el botón de encendido);
- sus ICs inhibidores no se hayan unido a sus ligandos (no se haya pulsado el botón de apagado);
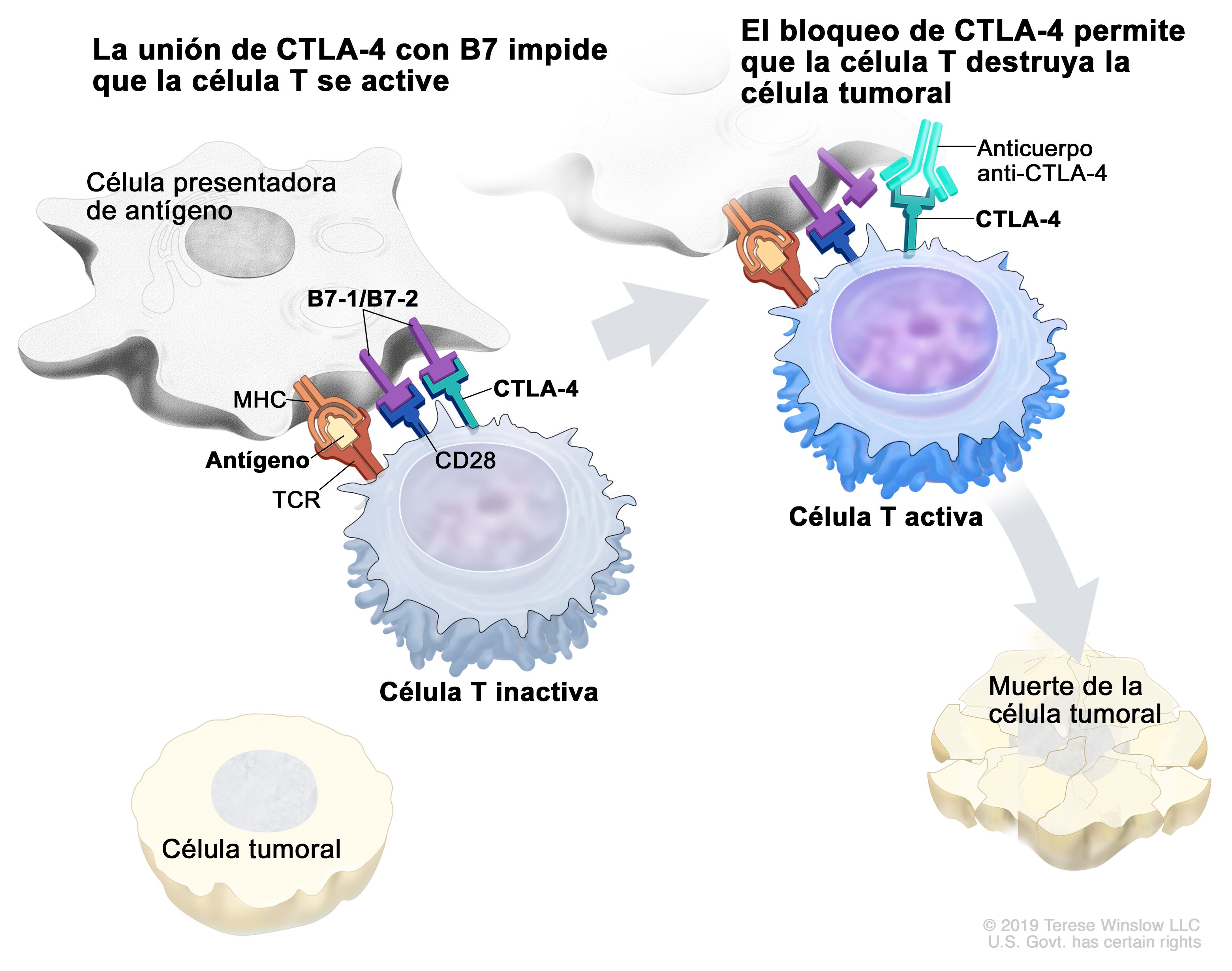
La presencia de ICs en la superficie de los linfocitos T citotóxicos y auxiliares es normal y permite que su actividad sea atenuada de forma natural cuando sea necesario...
- para evitar procesos autoinmunitarios: las propias células presentadoras de antígenos, cuando lo que presentan a un linfocito Th es un autoantígeno, pulsan sus ICs inhibidores;
- cuando ya se ha acabado con la amenaza: los linfocitos T supresores (T reguladores, Treg, subgrupo de T-CD4) desactivan a los linfocitos T citotóxicos y auxiliares que vencieron en la batalla, pulsando sus ICs inhibidores.
El problema es que las células tumorales se aprovechan de esto y tienen en su superficie moléculas ("ligandos") que se unen a los ICs inhibidores, activándolos (pulsan el interruptor de apagado). Los linfocitos T ya no actúan contra el tumor, sino que se alejan de él y quedan "agotados". El ligando PD-L1 pulsa el IC PD-1; el ligando B7 pulsa el IC CTLA-4; ambos "apagan" a los linfocitos T.
Enfoques Terapéuticos
Perfil genético
Es un análisis parcial del genotipo de las células tumorales, en las que se buscan mutaciones específicas en genes específicos que codifiquen para proteínas anómalas ya conocidas y que sirven como dianas de un tratamiento ya existente o en desarrollo.
Para hacerlo se puede buscar la presencia de esas mutaciones directamente (en el ADN, el genotipo) o indirectamente (en el ARN mensajero que expresan, llamado en conjunto transcriptoma).
Como los tumores FLC son de crecimiento lento y tienen poca carga mutacional (TMB), estos análisis suelen ser inútiles.
Silenciamiento de genes
Son terapias experimentales de uso en cánceres en los que el comportamiento tumoral de sus células es causado por un único gen defectuoso de secuencia de nucleótidos conocida y con una porción exclusiva (no presente en ningún otro lugar del genotipo de esa persona), la cual sirve de diana. Pero sirve también contra cualquier enfermedad hereditaria que se pueda curar simplemente silenciando a un único gen.
CRISPR: gene-knockout
CRISPR-Cas9 es un complejo nucleoproteínico con 2 componentes: una endonucleasa (una enzima capaz de hacer cortes en el interior de una molécula de ADN) y un ARN guía de 20 nucleótidos de longitud, que le dice a la nucleasa dónde cortar.
Tras el corte la célula introduce una secuencia de pares de nucleótidos al azar en el punto de corte y lo sella. En cualquier caso, si el corte se ha hecho dentro de un exón de un gen que codifica para una parte relevante de su proteína, todas las unidades de esta que se fabriquen a partir de ese momento no serían ya funcionales. Como se ha inactivado por completo la funcionalidad de un gen, se habla de gene-knockout. Y si la proteína inactivada es esencial para la vida de la célula (como la kinasa A), esta morirá.
Ese ARN guía se puede diseñar a medida para que CRISPR corte en el lugar indicado del genotipo de una persona, p.e. en el interior de un oncogen.
El problema en FLC es que la secuencia diana del oncogen es la zona de fusión de los 2 antiguos genes individuales y esta es un intrón (el que hay entre los exones 1 y 2). Y alterar un intrón carece de efecto en la proteína final, ya que los intrones son eliminados del ARNm antes de que este salga al citoplasma (maduración, corte y empalme, splicing).
Si se consiguiera sortear esta dificultad con las incesantes investigaciones para ampliar las capacidades de CRISPR, se conseguiría sin duda que el tumor se redujera y desapareciese, porque las células así modificadas mueren, ya que la proteína mutante, la kinasa A (hiperactiva o no), es necesaria para la vida de la célula.
Sería pues un tratamiento de una vez y definitivo. Y es idóneo para cualquier enfermedad debida a un único gen defectuoso, del que se puede prescindir (porque no es necesario o porque no lo son las células que lo portan, ya que hay otras similares no defectuosas), como en el FLC o la anemia falciforme.
ARNi: gene-knockdown
El ARN de interferencia (ARNi o ARNsi) es un fragmento pequeño de ARN que sirve para pegarse por complementariedad de bases al ARN mensajero (ARNm) maduro (sin intrones) codificado por un gen concreto (el oncogen, en este caso) e inactivarlo. Es decir, el oncogen se puede expresar transcribiéndose a ARNm, pero éste es "placado" por el ARNi antes de que llegue a los ribosomas y no puede traducirse a proteínas. De ello resulta que la "oncoproteína" deja de fabricarse... hasta cierto punto, porque es casi imposible que el ARNi consiga placar a tiempo a todas las cadenas de ARNm emitidas por gen mutante, todo el tiempo. Por eso se habla de gene-knockdown.
Previamente habría que crear en laboratorio el ARNi que placase sólo a los ARNm emitidos por el oncogen DNAJB1-PRKACA. Esto parece fácil, ya que esos ARNm tienen una región específica: la zona de fusión de los 2 antiguos genes individuales (exones 1 y 2, que en el ARNm maduro no están separados por ningún intrón): en esa región se encontraría la secuencia de nucleótidos diana. No obstante el gen DNAJB1-PRKACA, como todos los genes, presenta variaciones entre las personas, y podría haber diferencias en algún nucleótido en esa secuencia diana, por lo que el ARNi que funciona para una persona podría no funcionar para otra.
Igualmente, el ARNi se puede pegar al ARNm formando bucles, con lo cual no es estrictamente específico de su secuencia complementaria: puede así desactivar a más clases de ARNm, lo que es una fuente de posibles efectos secundarios de la que carece CRISPR.
Otro requisito es conseguir introducir el ARNi en las células. Se puede hacer metiéndolo dentro de liposomas que lo introducirán en la célula por endocitosis, como se hace en las vacunas de ARN. Pero también usando receptores de superficie de células concretas, entre ellas las tumorales (o de ser posible, sólo las tumorales).
Un receptor de superficie que permite el paso del ARNsi se llama ASGR1 y es específico de los hepatocitos, con lo que la terapia no podría interferir con ningún otro tipo de célula. La llave que lo abre se llama GalNAc y es a ella a la que hay que unir (conjugar) el ARNi para que lo cuele dentro de la célula.
El procedimiento se ha demostrado con FLC en este artículo de 2023-11 por el equipo de Sanford Simon (el padre de Elana y descubridor de la causa del FLC), que habla de muy buenos resultados en muestras de células tumorales extraídas de un paciente y cultivadas tanto in vitro como in vivo (xenoinjerto en ratones): se conseguía siempre meter el ARNsi fácilmente en las células tumorales, se frenaba la producción de la kinasa A mutante, los tumores se estancaban o menguaban de forma duradera y los ratones no padecían de toxicidad alguna. Pero como se trata de gene-knockdown, hay aún una expresión residual del oncogen y los tumores no eran eliminados. Por ello están intentando mejorarlo. Este otro artículo comenta y explica el descubrimiento.
Esta terapia parece muy factible a corto o medio plazo, por su facilidad de diseño e implementación, y poco peligrosa, por su elevada especificidad y porque no modifica el genotipo. Pero deja al oncogen intacto, por lo que para que resulte una terapia definitiva habría de tener una duración y ser de una intensidad tal que se bloqueen (casi) todos los ARNm de DNAJB1-PRKACA durante el tiempo suficiente como para que la ausencia de kinasa A haga morir a las células tumorales.
PROTAC
Las quimeras marcadoras de proteólisis (PROTACs) son el equivalente de CRIPSPR/Cas-9 a nivel proteínico. Son proteínas que marcan a otras proteínas para ser degradadas, de forma específica.
Tienen 2 partes: una específica, que se une a una clase concreta de proteínas diana; otra genérica, una ligasa que fija una cadena de ubiquitinas a la proteína diana, la cual queda marcada para su degradación en unos corpúsculos celulares llamados proteosomas.
El equipo de Sanford Simon está trabajando en el desarrollo de un PROTAC que degrade específicamente la enzima DNAJB1-PRKACA.
Posibles dificultades:
- DNAJB1-PRKACA, la proteína mutante con actividad kinasa que causa el FLC, es una fusión entre un fragmento pequeño de la proteína DNAJB1 (chaperona) y un fragmento grande de la proteína PRKACA (kinasa). Por consiguiente no ha de ser fácil diseñar un PROTAC que afecte sólo a la proteína de fusión y no a ninguna de las otras 2 en las células sanas. Para ello debería de tener su diana específicamente en la zona de fusión de DNAJB1-PRKACA. Esto ha de ser mucho más difícil de conseguir que un ARN de interferencia que tenga como diana la zona de fusión del ARNm mensajero que codifica para DNAJB1-PRKACA, pues en este caso todo lo que hay que hacer es encadenar una serie conocida de nucleótidos.
- Aún si se consigue un PROTAC que degrade específicamente la proteína DNAJB1-PRKACA y nada más, el oncogen, como en el caso del uso de ARNi, seguirá intacto. Por ello la terapia habría de tener igualmente una duración y ser de una intensidad tal, que se degraden (casi) todas las unidades de DNAJB1-PRKACA durante el tiempo suficiente como para que la ausencia de kinasa A haga morir a las células tumorales y resulte una terapia definitiva. De otro modo podría impedir la progresión de la enfermedad, pero nada más.
Quimioterapia
Inhibidores de PARP
La PARP o poly ADP-ribose polymerase es una enzima que identifica errores (típicamente delecciones) en el ADN tras su replicación (que sucede un poco antes de que la célula se divida por mitosis). PARP identifica la delección y la rellena de forma temporal con una cadena larga del nucleótido ADP. Esto sirve para señalar a las enzimas que reparan el ADN que hay que reparar esa zona, sustituyendo la cadena provisional de poli-ADP por los nucleótidos "correctos" que había antes de la delección.
Aunque parezca imposible, estas proteínas reparadoras lo que hacen es mirar qué nucleótidos hay en el mismo lugar del cromosoma homólogo, el que no ha sufrido la delección, y copiarlos en el lugar de la delección en sustitución de la cola de poli-A. Esto se llama "homologous recombination repair" (HRR). Si se ha perdido un gen del cromosoma 19 heredado del padre, miran la secuencia de nucleótidos que hay en el mismo lugar del cromosoma 19 heredado de la madre, y la copian en el del padre, en el lugar exacto, identificado por la presencia de la secuencia de poli-ADP.
Pero en el caso de pacientes con FLC la reparación HRR no es posible, quizás por ser la delección muy larga, y la acción de PARP, en realidad, es nociva, pues ayuda a prolongar la vida de la célula tumoral. De ahí que los inhibidores de PARP tengan actividad antitumoral en FLC y se hayan usado en combinación con vacunas de péptidos.
Inhibidores de kinasas
La proteína de fusión DNAJB1-PRKACA es la kinasa A codificada en condiciones normales por el gen PRKACA, pero que ahora es anormalmente hiperactiva. Más aún, todas las variantes de FLC parecen deberse a una kinasa A (mutante o no) hiperactiva en los hepatocitos que devienen tumorales. Es por eso que los inhibidores de kinasas (TKIs o mTOR-inhibitors) pueden ayudar contra el FLC.
Inhibidores de la replicación del ADN
Dificultan o inhiben la replicación del ADN (y a veces o en menor medida, también su transcripción), lo que impide la división de las células, lo que afecta más a las células tumorales que a las no tumorales. Por ello se usan como antitumorales, aunque serían menos útiles en FLC, de crecimiento lento. Tienen bastantes efectos secundarios, que pueden persistir tras el tratamiento.
El 5-fluorouracilo (5-FU) se usa en terapia combinada de FLC: nivolumab + 5-FU + interferon alfa-2b [ver terapias combinadas que usan ICIs].
Una terapia combinada de oxaliplatino, gemcitabina junto con lenvatinib ha sido objeto de un ensayo clínico: Gemcitabine-Oxaliplatin-Lenvatinib (GEMOX-LEN) for unresectable fibrolamellar carcinoma: Promising results in first 16 patients.
- Es de 2021-5.
- Enlace al póster.
- Presenta los resultados de un tratamiento quimioterapéutico triple con oxaliplatino, gemcitabina y lenvatinib.
- Se pudo evaluar a 16 pacientes, en general con cirugía y tratamientos previos.
- Los resultados son bastante buenos, con una tasa de control del tumor del 100%; ahora mismo no hay nada comparable:
- 7 con enfermedad controlada (reducción leve de los tumores, pero sin llegar al 30%).
- 8 con respuesta parcial (reducción de los tumores superior al 30% pero no total).
- 1 con remisión clínica (desaparición de los tumores).
Lenvatinib
Bloquea los receptores de la citoquina VEGF que hay en la superficie de las células endoteliales de los capilares. Como aquella estimula la angiogénesis, el levantinib la inhibe.
También bloquea los receptores de la citoquina FGFR, que estimula la proliferación de los fibroblastos, que a su vez ayudan a la proliferación y supervivencia de las células cancerosas.
Es al menos igual de útil que el sorafenib en tratamiento de carcinoma hepatocelular (HCC).
Efectos secundarios: hipertensión, fatiga, diarrea, nauseas, vómitos...
Antagonistas de la glutamina
Las células tumorales de FLC, de muchos otros tipos de cáncer, y las células con muy alta tasa de proliferación (leucocitos, enterocitos) consumen mucha glutamina (que utilizan como precursor metabólico de todo tipo de compuestos nitrogenados necesarios en procesos anabólicos) y cuando son privadas de ella no sólo no proliferan sino que mueren por apoptosis.
Al hacer esto, las células tumorales no sólo privan a los linfocitos T de la propia glutamina, sino que liberan como subproductos ácido láctico y amoniaco. Todo ello supone un microambiente químico hostil que impediría a los linfocitos T proliferar y sobrevivir cerca de ellas. Esto explicaría el bajo número de TILs en tumores FLC.
DON es un análogo de la glutamina que impide el uso de esta por muchos enzimas, con lo que las células tumorales mueren; pero también lo hacen las células del tracto gastrointestinal.
Por su parte DRP-104 (sirpiglenastat) es un compuesto inerte que es convertido a DON sólo en células tumorales, por lo que carece de sus efectos secundarios. Su empleo ha resultado en freno del crecimiento tumoral y restauración de la actividad de los linfocitos en ensayos de laboratorio.
El uso de DRP-104 combinado con un ICI anti-PDL1 en ratones dio como resultado una mejora de la supervivencia de estos y dio pie al inicio de un ensayo clínico.
Ensayos clínicos
DRP-104 in combination with durvalumab in patients with advanced stage fibrolamellar carcinoma:
- Fase I/II.
- De 2024-2 a 2027-2.
- Dr. Mark Yarchoan (Universidad Johns Hopkins, Baltimore).
- DRP-104 + Durvalumab (ICI anti-PDL1).
- Recluta: 12 años, Metástasis de FLC, Gen DNAJB1-PRKACA confirmado, Inmunoterapia previa inefectiva, No embarazada, No tratamientos anticancerosos 21 días antes del inicio, No terapias adicionales en los 21 días tras el inicio, No cirugías en los 28 días tras el inicio, No uso de corticoides, Tolerancia a anti-PD1 y anti-PDL1, No injertos ni transplantes, No autoinmunidad, No inmunodeficiencia, No HIV, No hepatitis B...
-
- Fase II/III.
- De 2018-5 a 2026-6.
- Ya no recluta.
- Este ensayo de fase II/III parcialmente aleatorizado estudia cómo de bien, en combinación con la cirugía, la quimioterapia combinada funciona en el tratamiento de niños y adultos jóvenes con hepatoblastoma o carcinoma hepatocelular. Los medicamentos utilizados en quimioterapia, como cisplatino, doxorrubicina, fluorouracilo, sulfato de vincristina, carboplatino, etopósido, irinotecán, sorafenib, gemcitabina y oxaliplatino, actúan de diferentes maneras para detener el crecimiento de las células tumorales, ya sea matándolas o impidiéndolas dividirse o evitando que se propaguen. Administrar quimioterapia combinada puede destruir más células tumorales que un tipo de quimioterapia solo.
Moduladores del sistema inmunitario
Interferón alfa
Es una citoquina que ayuda a desinhibir al sistema inmunitario en su lucha contra un tumor y que ayuda a que las propias células tumorales liberen citoquinas adicionales que atraigan a las células inmunitarias al tumor.
Anticuerpos monoclonales
Los anticuerpos monoclonales (MABs) son similares a los que tienen y liberan los linfocitos B, pero sintéticos. Se llaman monoclonales porque son idénticos, es decir, todos identifican al mismo antígeno.
En terapia antitumoral se pueden usar así:
- MABs que se unen y bloquean a proteínas de superficie necesarias para el desarrollo del tumor, pero no de las células tumorales; los 2 siguientes se usan como último recurso contra tumores de hígado:
- Bevacizumab bloquea a la citoquina VEGF, la cual induce la angiogénesis.
- Ramucirumab bloquea los receptores de VEGF que hay en la superficie de las células endoteliales de los capilares, inhibiendo también la angiogénesis.
- MABs que se unen y bloquean a proteínas de superficie de las células tumorales necesarias para el desarrollo del tumor:
- Trastuzumab bloquea la proteína de superficie HER2 de las células tumorales del cáncer de mama, las cuales dejan de crecer y proliferar.
- MABs que se unen a proteínas de superficie que sólo presenten las células tumorales (neoantígenos), con el objetivo de marcarlas para su posterior eliminación por los fagocitos, las células NK o las proteínas del complemento (los efectores de la respuesta inmune innata):
- Rituximab se une al neoantígeno CD20 de las células tumorales de los linfomas, facilitando su eliminación posterior por el S.I.
ICIs
Qué son
Los ICIs (inmune checkpoints inhibitors, inhibidores de los puntos de control inmunitarios) son anticuerpos monoclonales (MABs) cuya acción resulta en la desinhibición de los linfocitos T que reconocen a los neoantígenos de las células tumorales, a las que proceden ahora a eliminar.
Hay 2 clases:
- ICIs que se unen y bloquean a los ICs de tipo inhibidor de la superficie de los linfocitos T. Es como bloquear el interruptor de apagado que tienen los linfocitos T en su superficie, para que las células tumorales no puedan pulsarlo. Aparentemente serían de utilidad en multitud de tipos de tumores y de hecho se usan contra el FLC.
- Ipilimumab, Botensilimab: bloquean al IC llamado CTLA-4.
- Nivolumab, Pembrolizumab, Sintilimab, Balstilimab: bloquean al IC llamado PD-1. Sus efectos secundarios son: prurito, rash cutáneo, náuseas, tos, estreñimiento, dolor articular, diarrea y pérdida de apetito. Es objeto de un ensayo clínico: "Inhibición de puntos de control en el carcinoma hepatocelular pediátrico (incluido FLC)":
- Fase II.
- De 2020-11 a 2025-1.
- Dr. Allison O'Neill.
- Recluta.
- Pembrolizumab (ICI anti-PD1).
- ICIs que se unen y bloquean a los ligandos de la superficie de las células tumorales que se unen a los ICs de tipo inhibidor de la superficie de los linfocitos T. Es como bloquear el dedo con el que las células tumorales pulsan el interruptor de apagado que tienen los linfocitos T en su superficie. Igualmente serían de utilidad en multitud de tipos de tumores y se usan contra el FLC.
- Atezolizumab, Durvalumab bloquean al ligando del PD-1, es decir, al PD-L1, con idénticos efectos y eficacia que los bloqueadores del PD-1. Los efectos secundarios más frecuentes son perdida de apetito, diarrea, náuseas, vómitos, sensación de asfixia, fiebre, dolor en las articulaciones, cansancio y prurito. En ocasiones neumonitis, meningitis, pancreatitis, hipotiroidismo, hipertiroidismo, hepatitis, diabetes mellitus, insuficiencia suprarrenal y síndrome de Guillain-Barré.
Comentario
El uso de ICIs aislados contra FLC apenas es eficaz. Parece deberse a 2 motivos:
- La baja carga mutagénica (TMB) de los tumores FLC (una única proteína mutante, que además es recurrente) hace que se expresen pocos neoantígenos, lo que tiene poca capacidad de enfadar a los linfocitos; por tanto no bastaría con desinhibirlos: también habría que azuzarlos (p.e. con una vacuna de péptidos) o multiplicarlos artificialmente (p.e. con CAR-T).
- Las células tumorales de FLC no parecen expresar PD-L1.
- Se ha visto que cuando se inactiva una clase de ICs, el linfocito T produce más cantidad de otras clases.
El uso combinado de 2 ICIs parece ser más eficaz, pero tiene que ser frente a ICs diferentes: no tiene sentido usar un inhibidor de PD-1 junto con un inhibidor de PD-L1.
Los inhibidores de PD-1 parecen presentar igual eficacia que los inhibidores de PD-L1.
La terapia con un combinado de ICIs debería de ser útil en todos los casos de FLC que se deban a proteínas de fusión (sea DNAJB1-PRKACA, ATP1B1-PRKACA u otra) ya que estas sirven como neoantígenos reconocibles por los linfocitos T, pero probablemente no funcione cuando la causa sea la inactivación de un gen que codifique para un modulador de la kinasa A, ya que en este caso no habría nada que pudiera funcionar como neoantígeno. De ahí la importancia de confirmar la presencia de un gen de fusión en las células tumorales.
La terapia con ICIs puede generar reacciones autoinmunitarias significativas.
Terapias combinadas de ICIs
- Ipilimumab (ICI anti-CTLA4) + Nivolumab (ICI anti-PD1); aprobada para HCC.
- Botensilimab (ICI anti-CTLA4) + Balstilimab (ICI anti-PD1). Es objeto de un ensayo clínico: "Anticuerpo monoclonal anti-CTLA-4 en cáncer avanzado (con o sin anti-PD1)":
- Incluye una cohorte para FLC en la que se usa también Balstilimab (anti PD1).
- Fase I.
- De 2019-4 a 2026-12.
- Agenus Inc., Dr. Ghassan Abou-Alfa.
- 13 localizaciones en USA, 1 en Londres.
- Recluta: 18 años, cáncer avanzado, no embarazada, no terapias en las 3 semanas anteriores, Tolerancia a MABs, No injertos ni transplantes, No autoinmunidad, No inmunodeficiencia, No HIV, No hepatitis B o C...
Terapias combinadas que usan ICIs
- Nivolumab (ICI anti-PD1) + 5-FU (inhibidor de la mitosis) + interferon alfa-2b (estimulador del SI). Se está usando en pacientes con FLC. Es objeto de un ensayo clínico:
- Fase I/II.
- De 2021-1 a 2025-7.
- Dr. Sunyoung Lee (University of Texas), Dr. Ahmed Kaseb (University of Texas).
- Ya no recluta.
- Atezolizumab (ICI anti-PDL1) + bevacizumab (inhibidor de la angiogénesis); aprobada para HCC.
- Nivolumab (ICI anti-PD1) + lenvatinib (inhibidor de la angiogénesis).
- Vacunas de péptidos + ICIs [ver abajo].
Vacunas de péptidos
Objetivo
Conseguir que el paciente genere muchísimos linfocitos T específicos contra los neoantígenos del FLC.
Cómo funcionan
La proteína DNAJB1-PRKACA tiene una parte específica, que quizás no esté presente en ninguna otra proteína humana, que es la zona de fusión entre la proteína codificada por el gen DNAJB1 y la codificada por el gen PRKACA.
Es por ello que, conocida la secuencia de aminoácidos de la proteína DNAJB1-PRKACA y en particular de su zona de fusión, se pueden sintetizar en el laboratorio diversas cadenas cortas de aminoácidos (péptidos) que comprendan el punto de fusión (p.e. con 10 aminoácidos de un lado y 10 de otro) para que funcionen como antígenos inyectados en vacunas.
Esto hará que el clon de linfocitos T que sabe reconocer a ese antígeno se multiplique y multiplique sus receptores de superficie (TCRs), lo que puede dar lugar a...
- una respuesta inmunitaria masiva que avasalle a las células tumorales, que no serán ya capaces de "apagarlos" a todos manipulando sus puntos de control (ICs);
- linfocitos de memoria específicos contra FLC que acabarían con cualquier intento de recidiva.
Ensayos clínicos
Estas vacunas sólo están disponibles hoy en día como parte de ensayos clínicos en los que se combinan con ICIs, ya que no parece probable que por muchos que sean los linfocitos T anti-FLC puedan evitar que el tumor "los apague":
- Universidad Johns Hopkins, Baltimore:
- Fase I.
- De 2020-4 a 2027-3.
- Dr. Mark Yarchoan.
- Vacuna (con péptido de 24 aminoácidos, 12 de cada lado del punto de fusión) + Nivolumab (ICI anti-PD1) + Ipilimumab (ICI anti-CTLA4).
- El tratamiento con nivolumab + ipilimumab ya ha funcionado por sí solo, sin ayuda de vacunas.
- Recluta: Pacientes con metástasis de FLC, Gen DNAJB1-PRKACA confirmado, No embarazada, No tratamientos anticancerosos 14 días antes del inicio, No cirugías ni tratamientos anticancerosos ni vacunas antimicrobianas ni esteroides en los 28 días tras el inicio, Tolerancia a MABs, No injertos ni transplantes, No autoinmunidad, No inmunodeficiencia, No HIV, No hepatitis B o C...
- Cohorte A: 12 años. Sin tratamiento previo con ICIs.
- Cohorte B: 12 años. Con tratamiento previo con ICIs, siempre que no haya generado alta toxicidad.
- Cohorte C: 18 años. Pacientes con tumores sólidos adicionales no de FLC sin tratamiento previo con ICIs.
- Resultados: el 75% mostró respuesta inmunitaria:
- 50% : al menos varios meses sin crecimiento tumoral.
- 25%: decrecimiento dramático del tumor; libres de cáncer durante 3 a 5 años.
- Hospital Universitario de Tubinga:
- Fase I.
- De 2023-6 a 2027-1.
- Dr. Salih, Dra. Juliane Walz.
- Vacuna FusionVAC-22 (con péptido de 24 aminoácidos, 12 de cada lado del punto de fusión) + atezolizumab (ICI de tipo anti-PDL1).
- Recluta: 18 años, Pacientes con metástasis de FLC (o sin ellos si no hay una terapia estándar, esta es inútil o no se puede usar), Gen DNAJB1-PRKACA confirmado, No embarazada, No participación en tests clínicos similares 30 días antes del inicio, No tratamientos anticancerosos 14 días antes del inicio, No tratamientos con corticoides 8 días antes del inicio, No cirugías ni tratamientos inmunoterapéuticos ni vacunas con m.o. vivos en los 28 días tras el inicio, Tolerancia a MABs, No injertos ni transplantes, No autoinmunidad, No inmunodeficiencia, No covid, No HIV, No hepatitis B o C...
- Hospital Universitario de Tubinga:
- Fase I.
- De 2025-6 a 2028-11.
- Dr. Salih, Dra. Juliane Walz.
- Vacuna FusionVAC-22 (con péptido de 24 aminoácidos, 12 de cada lado del punto de fusión).
- Es un estudio de la eficacia de la vacuna de péptido para prevenir la reaparición del cáncer en pacientes operados.
Bacterias con neoantígenos
Objetivo
Conseguir que el paciente genere muchísimos linfocitos T específicos contra los neoantígenos del FLC.
Cómo funciona
- Se identifica el gen que codifique para un neoantígeno tumoral.
- Se clona en bacterias, para que estas expresen el neoantígeno.
- Las bacterias transgénicas son presentadas a linfocitos T auxiliares extraídos del paciente, que se activan contra ese neoantígeno, probablemente mucho más que con una vacuna consistente en fragmentos del neoantígeno.
- Una vez reintroducidos en el cuerpo del paciente, linfocitos T auxiliares activados buscan a los linfocitos T citotóxicos para activarlos también.
- Los T citotóxicos activados eliminan el tumor y algunos permanecen como células de memoria.
Estado
- Terapia experimental, sin tests clínicos.
- Dr. Esteban Veiga, del CNB-CSIC.
CAR-T
Objetivo
Conseguir que el paciente adquiera muchísimos linfocitos T especialmente eficaces en la detección de los neoantígenos de las células tumorales y en la eliminación de estas.
Cómo funciona
- Se identifica un gen que codifique para un la región variable de un anticuerpo que identifique de forma eficaz a un neoantígeno de las células tumorales (en lugar de un TCR, ya que los anticuerpos no necesitan que los antígenos les sean presentados por moléculas HLA).
- Por ingeniería genética, se combina con el gen de un IC estimulador (p.e. CD28).
- Este gen creado por ingenieria genética codificará para una proteína quimérica (CAR) que:
- Es capaz de reconocer a un tipo de neoantígenos de las células tumorales;
- Le da igual cuál de las muchas clases de proteínas HLA presentes en la célula tumoral le presente el neoantígeno;
- Hace innecesario que a la célula T que lo porte se le pulse el interruptor de encendido (que la célula tumoral tenga ligandos complementarios de sus ICs de tipo estimulador).
- Se extraen leucocitos de la sangre paciente (2-3 horas) y se seleccionan los linfocitos T.
- Se transfiere el gen sintético a los linfocitos T con ayuda de un vector (p.e. un virus atenuado).
- Los linfocitos T transgénicos (CAR-T) se hacen proliferar en cultivos de laboratorio (semanas).
- Y se transfieren masivamente al paciente, dando lugar a...
- un gran número de linfocitos T capaces de detectar y matar a las células tumorales;.
- linfocitos de memoria específicos contra FLC que acabarían con cualquier intento de recidiva.
Saber más
- Terapia de células CAR-T y sus efectos secundarios
- ¿Qué es la terapia CAR-T?
- Terapia con células CAR-T. De la Clínica Universitaria de Navarra, que ya lo está aplicando en varios tipos de cáncer.
Dificultades
- CAR-T funciona muy bien para tumores líquidos (p.e. linfomas), pero funciona mal contra los tumores sólidos, posiblemente porque en ellos se genera un microambiente que causa el agotamiento de los linfocitos inducido por la manipulación que las células tumorales hacen de los ICs. Por ello quizás sólo tenga sentido usar esta terapia contra el FLC en combinación con ICIs, igual que las vacunas de péptidos o cualquier otra terapia basada en aumentar los linfocitos T específicos.
El artículo "Prospects and challenges of CAR-T cell therapy combined with ICIs" hace un buen repaso de la situación actual:- Es de 2024-3.
- Muestra cómo la terapia CAR-T funciona con frecuencia mucho mejor en tumores sólidos si es combinada con ICIs, llegándose a casos de remisión completa del cáncer; en otros casos (cáncer de pulmón) el efecto es el contrario; y en otros no se alcanza el objetivo por los elevados efectos secundarios que a veces tienen los ICIs.
- Muestra cómo los linfocitos CAR-T modificados adicionalmente para que ellos mismos produzcan y liberen ICIs anti-PD1 en el lugar del tumor (linfocitos CAR-T PD1-silenciados), funcionan bien por sí solos en tumores sólidos y suponen una alternativa con muchos menos efectos secundarios.
- Muestra cómo los linfocitos CAR-T modificados adicionalmente para carecer de ICs PD-1 funcionan igualmente bien por sí solos en tumores sólidos (incluido el carcinoma hepatocelular) al no poder ser "apagados" por los ligandos PD-L1 de las células tumorales. Sin embargo estos linfocitos CAR-T PD1-KO no pueden reproducirse, a diferencia de los linfocitos CAR-T PD1-silenciados, por lo que hay dudas sobre cuál de las 2 alternativas es mejor.
- Muestra cómo los linfocitos CAR-T modificados adicionalmente para carecer de receptores CTLA-4 (linfocitos CAR-T CTLA4-KO) funcionan aún mejor que los anteriores y sí pueden reproducirse.
- Muestra cómo los linfocitos CAR-T modificados adicionalmente para carecer de varios ICs inhibidores (p.e. CTLA-4 + PD-1) funcionan aún mejor que los anteriores.
- Muestra cómo la técnica usada para eliminar los genes de los ICs y añadir el gen CAR influye mucho en la eficacia final del CAR-T; las últimas tecnologías de producción de CAR-T alcanzan tasas de eliminación total de los tumores muy altas.
- Muestra una alternativa más light que reduce mucho todo tipo de riesgos (p.e. autoinmunidad) porque no se llega a tocar el genoma del linfocito T: en vez de añadir el gen de CAR se añade su ARNm; y en vez de romper el gen del IC, se silencia con ARNi; los ARN se transportan al linfocito T no con un virus atenuado (ya no hace falta y siempre es arriesgado) sino con un liposoma (como en las vacunas de ARN); ahora no quedarán células de memoria, pero podría no hacer falta, y además un linfocito T que no se pueda inhibir jamás por carecer de ICs puede ser peligroso.
- CAR-T tiene efectos secundarios que son tanto mayores cuanto mayor es el tamaño de los tumores en el momento del tratamiento: es la tormenta de citoquinas, que si es muy grande puede llegar a causar daños serios en órganos, incluido en encéfalo.
- Una terapia con CAR-T es muy cara: cuesta entre 500.000 y 1.000.000 $ en USA.
Selección de TILs
Se extraen linfocitos de uno de los tumores del paciente. Se identifican aquellos activos contra las células cancerígenas. Se multiplican en un laboratorio. Se transfunden en un mayor número a la paciente para atacar de este modo su tumor.
Se ha utilizado en 2018 para sanar por completo a una mujer de 49 años con cáncer de mama metastásico, completamente desahuciada.
Tiene muy buena pinta. Pero es totalmente personalizada, por lo que quizás sea cara.
Transplante hepático
- Es posible cuando no ha habido metástasis.
- Puede ser ser un donante vivo sano, cuyo hígado se regenerará. No es necesario que sea de un familiar.
- Vida normal tras 2 meses, recuperación completa lleva hasta 6 meses.
- Medicación anti-rechazo de por vida.
- Es fácil que el transplantado sea inelegible para un tratamiento inmunoterapéutico futuro, si el cáncer reaparece.
Biomarcadores
SOD3
La Superóxido Dismutasa 3 (SOD3) es una enzima que tenemos en el plasma intercelular de muchos tejidos y que lo limpia del ión superóxido (un radical libre útil en la lucha contra patógenos, pero que favorece el cáncer, la diabetes o el Alzheimer). Y por ello favorece la salida de los linfocitos desde la sangre a los tejidos, ya que estos son inhibidos por aquel.
Por eso, en la mayoría de los tumores hay un silenciamiento de SOD3, de forma que sus niveles son muy bajos. Y por eso sirve como biomarcador de la posible evolución de un cáncer, ya que cuanto mayores sean sus niveles, mayor será la infiltración inmunitaria en el tumor y mejor será el pronóstico.
Esta correlación positiva entre niveles altos de SOD3, niveles altos de TILs y buen pronóstico se ha comprobado en una cohorte de 95 pacientes de cáncer colorrectal.
Según Santos Mañes, del CNB-CSIC: “Nosotros observamos que el 95% de los pacientes con niveles altos de SOD3 en sus tumores no volvían a padecer la enfermedad tras someterse a cirugía, mientras que un 40% de los que tenían niveles bajos de SOD3 sufrieron una recidiva del tumor antes de pasar cinco años tras la cirugía. La supervivencia era mayor en los primeros pacientes”.
Igualmente debería de ser un buen indicador del beneficio de una terapia con ICIs: estos sirven de poco si hay pocos TILs a los que desinhibir.
Enlaces Útiles
Artículos de investigación sobre terapias de FLC, ordenados de más reciente a más antiguo, de la National Library of Medicine. Se actualiza continuamente (hay que refrescar la página).
Fibrolamellar Cancer Foundation. Sitio web de la Fibrolamellar Cancer Foundation que informa de tratamientos, investigación destacada y ensayos clínicos actuales.
Perspectives on FLC immunotherapy. Artículo de 2024-5 de la FCF que resume los últimos avances en inmunoterapia.